Are you ready for Brexit? Do you have a Brexit contingency plan?
You may need either an EU/EC European authorized representative based in EU-27 countries or a UK authorised representative (so-called "UK Responsible Person") based in UK, or may even need both EU & UK representatives, depending on different brexit scenarios.
Register/Notify your MD-Medical Devices & IVD-In Vitro Diagnostic Medical
Devices with MHRA in UK & other EEA (EU/EFTA) authorities by world-leading
consultancy- Wellkang team based in both UK (England) & EU-27 (Ireland).
Wellkang team can help you under all Brexit scenarios!
Click here to get FREE Guide Now! |
Steps for Class III medical devices compliance
- Classification: ensure the device is a Class III medical device.
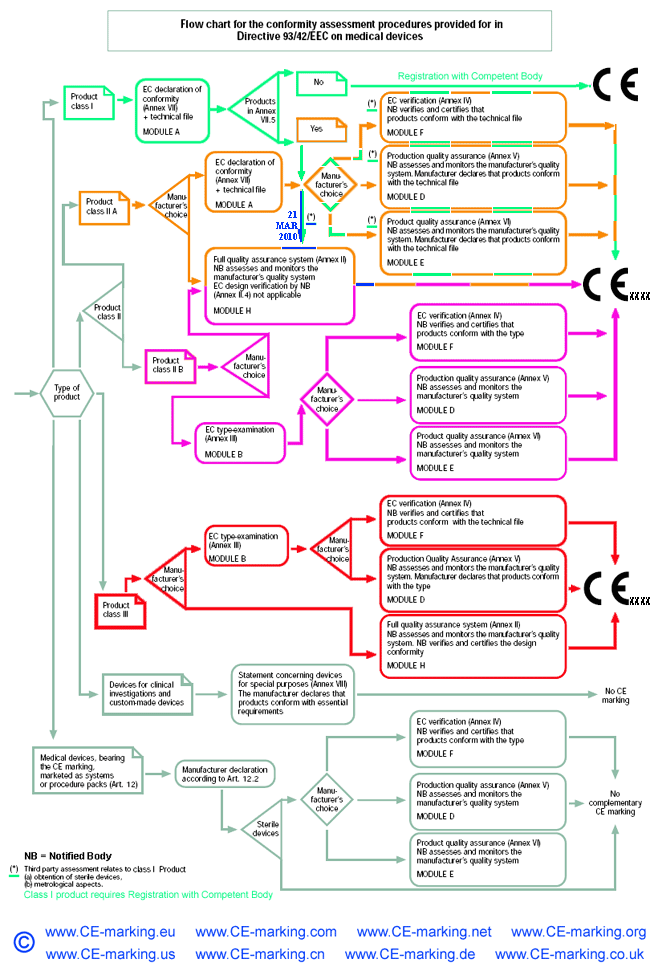
- Choose Conformity Assessment Route: refer the flow chart below.
- Compile the Technical File.
- Obtain certification from a Notified Body
- Declaration of Conformity.
- Appoint an Authorised Representative. (Hold the Tech Files for inspection by the Competent Authority)
- Vigilance and Post Market Surveillance. (affix CE marking & market the products)
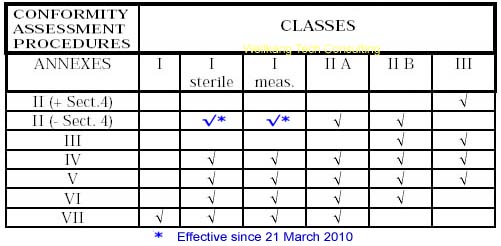
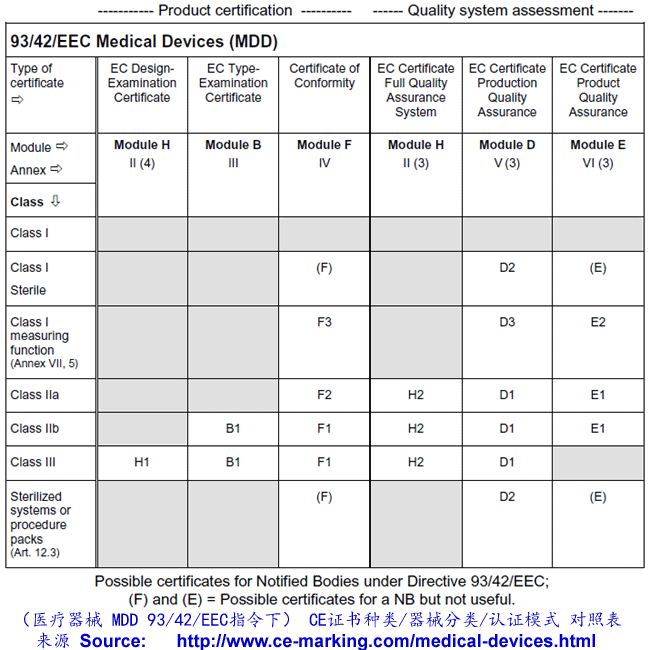
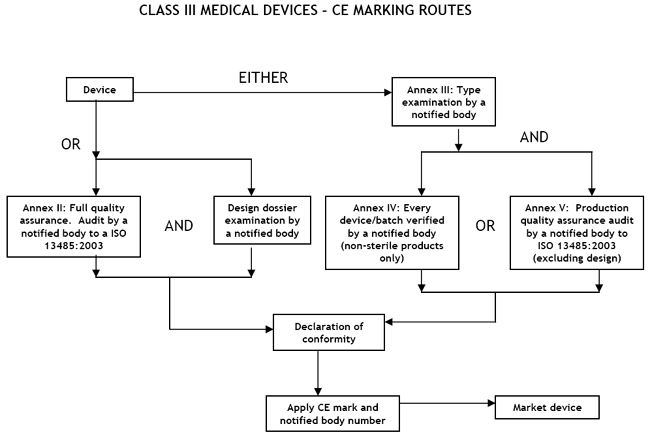
Class III Medical Devices: Conformity Assessment Routes
The conformity assessment routes for Class III Medical Devices
In the case of devices falling within Class III, other than devices
which are custom-made or intended for clinical investigations, the
manufacturer shall, in order to affix the CE marking, either:
- follow the procedure relating to the EC declaration of conformity
set out in Annex II (full quality assurance);
or
- follow the procedure relating to the EC type-examination set out in
Annex III, coupled with:
- (i) the procedure relating to the EC verification set out in Annex
IV;
or
- (ii) the procedure relating to the EC declaration of conformity set
out in Annex V (production quality assurance).
Class III controls are similar to those for Class IIb devices but additionally require the
manufacturer to submit the design dossier to the Notified Body for approval under Annex
II and do not allow the Annex III/Annex VI option.
There are two routes:
- a Notified Body must carry out either an Annex II audit of the full
quality assurance system (ISO 13485:2003), plus that the manufacturer must submit the design dossier to the Notified Body for approval under Annex
II, or
- a type-examination (Annex III) plus one
of the two options given here:
- Examination and testing of each product or homogenous batch of products (Annex IV);
or
- Audit of the production quality assurance system (Annex V:) ISO 13485:2003 (excluding
Design)
Once the manufacturer has received certification from the Notified Body he may CE mark his
products and place them on the market.

FAQ/Q&A: Questions and Answers about CE Marking of Medical Devices
About CE Marking:
| Home | About Us | Contact Us | Copyright & Disclaimer | Privacy | Log-in | Order Now | |